Definitie en symptomen
Polycytemia vera is onderdeel van een groep ziekten die myeloproliferatieve aandoeningen worden genoemd. Hieronder vallen de volgende ziektebeelden: polycytemia vera, essentiële trombocytemie en myelofibrose. Bij de meeste patiënten is er duidelijk sprake van een van deze drie beelden, maar er zijn ook patiënten bij wie het van meet af aan moeilijk is de aandoening in te delen. Bij hen worden zogenaamde ‘mengbeelden’ gezien met kenmerken van twee (of meer) verschillende groepen. Ook kan het voorkomen dat het ene ziektebeeld overgaat in het andere ziektebeeld. We gaan hier nu alleen in op het ziektebeeld polycytemia vera.
Polycytemia vera (letterlijk: ‘de ware ziekte met een teveel aan bloed’) is een ziekte waarbij sprake is van een ongecontroleerde toename van rode bloedcellen. De oorzaak van de toegenomen aanmaak ligt in de voorlopercel, de stamcel verantwoordelijk voor de bloedcelaanmaak (zie dia 2). In die stamcel zijn afwijkingen ontstaan (de zogenaamde JAK2-mutatie) waardoor de stamcel al gaat delen zonder daar de nodige signalen voor ontvangen te hebben (dia 3 en 4). De stamcel rijpt niet alleen uit tot rode cellen, maar ook tot vele andere bloedelementen, waarvan een deel extra mee gaat groeien (dia 3). Daardoor zijn niet alleen de rode bloedcellen verhoogd, maar meestal ook de witte cellen en de bloedplaatjes.

Dia 1
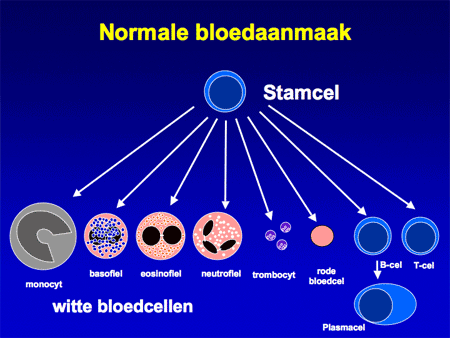
Dia 2
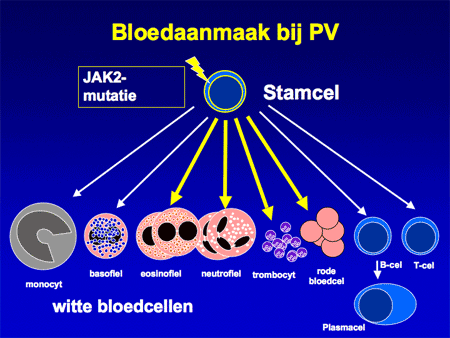
Dia 3
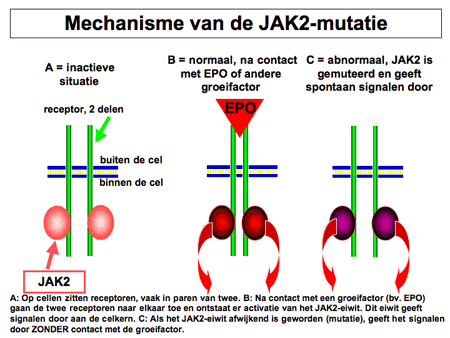
Dia 4
Door de toename aan rode cellen is het hemoglobinegehalte (Hb) te hoog en heeft de patiënt een opvallend rode kleur. Hoewel dit ogenschijnlijk gezond lijkt, is dit niet zo. Het hoge bloedgehalte maakt het bloed stroperig, het gaat langzamer stromen, waardoor er een groot risico op stolling (trombose) ontstaat in de aderen, maar ook in de slagaderen. Dit betekent dat er ook een toegenomen kans is op een hartinfarct, herseninfarct of inwendige trombose van de buikaderen. De kans op trombose wordt groter naarmate een patiënt ouder is en ook het aantal bloedplaatjes mede verhoogd is. Patiënten met een PV hebben vaak symptomen zoals hoofdpijn of moeite met scherp zien, hevige jeuk na het douchen en – heel opvallend – soms ernstige pijn in de voeten of vingers, waarbij dan een rood-blauwe verkleuring is te zien. Deze pijn heet erytromelalgie. Soms is er naast de trombose ook sprake van een verhoogde bloedingsneiging, omdat de bloedplaatjes niet meer goed werken wanneer ze sterk verhoogd zijn. Bij die bloedingsneiging kunnen patiënten inwendige bloedingen krijgen van maag of darmen. Een deel van de patiënten heeft een vergrote milt.
De toevoeging ‘vera’, ‘de ware’, geeft meteen aan dat er veel vormen van polycytemie zijn die een andere oorzaak hebben. Dat is ook belangrijk om te weten: het merendeel van de patiënten met een ogenschijnlijk teveel aan rode bloedcellen heeft dit door meestal onschuldige oorzaken, zonder dat er sprake is van een ongecontroleerde toename (dia 5). Deze toename is goed te begrijpen vanuit de functie van rode bloedcellen: zuurstoftransport. Het lichaam maakt bij een tekort aan zuurstof extra rode bloedcellen aan. Voorbeelden van een ‘onschuldig’ verhoogd gehalte aan rode bloedcellen is de polycytemie bij mensen die hoog in de bergen wonen: bij hen is een aanpassing opgetreden door de ijlere lucht, waar immers minder zuurstof in zit. Polycytemie wordt ook gezien bij forse rokers, want door de chronische koolmonoxidevergiftiging denkt het lichaam te weinig zuurstof beschikbaar te hebben, waardoor extra rode cellen aangemaakt worden. Een compensatoire verhoging van het bloedgehalte komt ook voor bij patiënten met een hart- of longaandoening of met ernstig overgewicht. Een modern voorbeeld van polycytemie is het hoge hemoglobinegehalte dat ontstaat na misbruik van Epo (erytropoietine) in de wielersport. Ten slotte is er ook nog een groep patiënten die een ogenschijnlijk verhoogd hemoglobinegehalte heeft, maar waar bij verder meten sprake is van een pseudo-verhoging door andere oorzaken, zoals stress of uitdroging.

Dia 5
Onderzoek gericht op polycytemia vera
Om de diagnose polycytemia vera te stellen is het nodig aanvullend onderzoek te doen om de echte PV te onderscheiden van de vele andere vormen van polycytemie (dia 6). Bij dit onderzoek is het belangrijk dat er vragen gesteld worden en het lichamelijk onderzoek gericht is op aandoeningen die met zuurstoftekort gepaard gaan. Daarnaast moet specifiek aandacht worden besteed aan risicofactoren voor hart- en vaatziekten.
Bij het bloedonderzoek wordt allereerst het bloedbeeld goed in kaart gebracht, wordt de JAK2-mutatie bepaald, misschien ook het erytropoietinegehalte, en verder factoren die belangrijk zijn voor de zuurstofgaswisseling, hart- en vaatziekten en eventuele andere relevante zaken. Bij twijfel wordt een zogenaamde ‘ery-volume’-meting gedaan, waarbij de echte polycytemie kan worden onderscheiden van de pseudo-polycytemie. Vrijwel steeds zal het ook nodig zijn een beenmergonderzoek te doen. Hiermee kan vaak heel goed onderscheid gemaakt worden tussen een compensatoire toename van bloedcellen of een ongecontroleerde toename. Naast een analyse op de JAK2-mutatie moet ook naar de chromosomen worden gekeken door middel van cytogenetisch onderzoek (zie chromosomenonderzoek). Tot slot wordt (althans in het UMCG) het beenmerg in kweek gebracht om te zien of de groei van rode cellen inderdaad ongecontroleerd is. In dit geval zullen de stamcellen uitgroeien tot celkolonies (dia 7) zonder dat het bloedhormoon erytropoietine toegevoegd hoeft te worden.

Dia 6

Dia 7
Behandeling van polycytemia vera
Het voornaamste doel van de behandeling is het voorkomen van complicaties (dia 8 en 9). Dit betekent dat het niet voldoende is om alleen het bloedgehalte naar beneden te krijgen, maar dat daarbij ook actief moet worden geprobeerd de kansen op het krijgen van trombose, een bloeding of een hart- of vaatziekte te verkleinen. De behandeling van PV is dus breder dan alleen gericht op het bloed. Ook de medische voorgeschiedenis (al eerder trombose of een hart- of vaatziekte gehad, of de aanwezigheid van risicofactoren zoals hoge bloeddruk, suikerziekte, roken, verhoogd cholesterolgehalte) is belangrijk voor het te bepalen beleid.

Dia 8

Dia 9
Alle patiënten worden behandeld met Ascal, dat de werking van de bloedplaatjes remt en zo de kans op trombose in aderen en slagaderen vermindert. Rokers moeten pertinent stoppen met roken, de bloeddruk moet goed gereguleerd zijn en zo nodig moet het cholesterolgehalte van het bloed met behulp van medicatie verlaagd worden.
Daarnaast wordt door middel van aderlatingen begonnen met het verlagen van het bloedgehalte. Bij een aderlating wordt 300 tot 500 ml bloed afgetapt, op precies dezelfde manier zoals dat bij donoren voor een bloedtransfusie gedaan wordt. De aderlatingen kunnen plaatsvinden met verschillende intensiteit. Gestreefd moet worden naar een hematocriet (Ht) lager dan 0,45 bij mannen en lager dan 0,42 bij vrouwen. Aanvankelijk kan om de dag of twee keer per week adergelaten worden. Nadat het streefniveau is bereikt, is meestal een onderhoudsbehandeling nodig van één keer per één tot drie maanden.
Bij ouderen is aderlaten meestal niet voldoende en wordt geadviseerd celremmende medicatie toe te voegen die de aanmaak van rode cellen actief remt. Zo’n middel is hydroxyureum, dat in capsules kan worden ingenomen en dat door de meerderheid van de patiënten zonder enige bijwerking wordt verdragen. Omdat hydroxyureum tot de chemotherapie gerekend wordt, krijgen jongere patiënten als alternatief therapie met interferon alfa aangeboden, dat per zelfinjectie toegediend moet worden. Interferon zou ook beter werken tegen de jeuk, maar heeft ook veel bijwerkingen zoals moeheid, spierpijn en soms zelfs een ernstig sombere stemming. Een alternatief voor de jeuk is het middel paroxetine. Een laatste alternatief is een behandeling met radioactief fosfor, dat vooral geschikt is voor patiënten ouder dan 70 jaar bij wie de andere behandelingen problemen geven.
Wat te doen bij operaties?
Patiënten met een PV mogen niet geopereerd worden zolang zij nog niet behandeld zijn. De regel is dat het bloed minstens drie maanden op het goede lage niveau gebracht moet zijn voordat de chirurg aan het werk mag gaan. Gebeurt dit niet, dan is het risico op complicaties tijdens en direct na de operaties erg groot. Soms is er geen keuze en zal het gaan om een spoedoperatie, maar als er sprake is van een geplande operatie (zoals een nieuwe heup, liesbreukcorrectie en dergelijke), moet deze bij voorkeur uitgesteld worden.
Toekomstverwachting
Patiënten met een PV hebben een levensverwachting van vele jaren, mits ze goed behandeld worden voor de ziekte zelf en mits de risicofactoren voor hart- en vaatziekten ook goed aangepakt worden. Bij sommige patiënten zal op de lange termijn, na zo’n vijftien tot twintig jaar, het beenmerg minder goed gaan functioneren en een verbindweefseling doormaken, waardoor uiteindelijk een tekort aan alle cellen zal ontstaan samen met een sterk vergrote milt. Het is niet goed bekend bij wie dit fenomeen zal optreden. Het lijkt erop dat hydroxyureum hiertegen beschermt.

Dia 10
Meer weten?
De MPN Stichting is een actieve patiëntenvereniging die brochures uitgeeft voor zowel patiënten als artsen. Daarnaast wordt veel aandacht besteed aan lotgenotencontact en belangenbehartiging. Het correspondentieadres is:
MPN Stichting
Postbus 10496
6000 GL Weert
Tel: 088-0074300
E-mail: info@mpn-stichting.nl
www.mpn-stichting.nl